肺脏位于胸腔内,是人体重要的呼吸器官,在呼吸过程中除了吸入氧气以外,空气中的微生物、有害颗粒等也会随着呼吸进入肺部,这些有害物质可造成肺部损伤,引发各类呼吸系统疾病,其中肺纤维化是多种间质性肺疾病的终末期肺部表现之一,现有的药物无法有效逆转纤维化进程,而促进纤维化肺组织再生有望恢复受损的肺泡和血管结构,为包括肺纤维化在内的多种疾病提供新的治疗策略。接下来,要介绍的这篇文献报道了TGM2(transglutaminase 2,谷氨酰胺转胺酶2)介导的TPI1(Triosephosphate isomerase 1,丙糖磷酸异构酶1)的多巴胺修饰可通过抑制铁死亡促进肺再生的新机制,这对开发抑制肺纤维化进程的治疗靶点具有重要的临床意义。在此研究中,作者使用了汉恒生物提供的VEC血清型+Tie内皮细胞特异性启动子的AAV病毒实现了肺部内皮细胞的特异性基因表达调控。

2024年8月6日,四川大学丁楅森、曹中炜团队和华西医院的叶庭洪、蒲强、张鸣和哈尔滨医科大学的杨力明作为共同通讯作者在《Cell Metabolism》(IF=27.7)发表题为“Dopaminylation of endothelial TPI1 suppresses ferroptotic angiocrine signals to promote lung regeneration over fibrosis ”的研究论文。该研究首先通过构建小鼠肺再生模型和肺内皮Tgm2特异性敲除小鼠并结合单细胞测序分析,发现肺内皮细胞TGM2介导的单胺化可抑制铁死亡进而维持肺再生,随后为研究其具体机制,作者通过体内外实验、化学蛋白质组学、点击化学等技术发现多巴胺能通过TGM2对内皮细胞中的TPI1的Q65位点进行多巴胺化修饰,此修饰可以促进DHAP(dihydroxyacetone phosphate,二羟丙酮磷酸)转化为GAP(glyceraldehyde 3-phosphate,3-磷酸甘油醛)的活性,从而抑制脂质过氧化和铁死亡,促进肺再生。最后,利用临床样本和肺纤维化小鼠模型证实了TPI1多巴胺化对肺纤维化的抑制作用,通过小分子药物pro-DA(propargylated dopamine,丙炔化多巴胺)恢复内皮TPI1多巴胺化水平,能抑制内皮细胞铁死亡,恢复促进再生的血管分泌因子的表达,从而减缓肺纤维化进程,为器官纤维化的治疗提供了新希望。
下面,我们一起来看看本文的主要研究结果吧:
首先,作者建立左肺切除手术(pneumonectomy,PNX)的小鼠模型并进行单细胞测序及WB检测,发现PNX小鼠的TGM2(单胺类神经递质,如多巴胺、血清素等可通过此酶共价修饰蛋白质,此过程称为单胺化) 在肺内皮细胞(Endothelial cell, EC)中选择性上调,提示TGM2依赖性单胺化可能在调节肺血管再生中起重要作用。为了验证这一假设,作者构建了肺EC特异性Tgm2敲除小鼠(Tgm2i∆EC/i∆EC),发现Tgm2i∆EC/i∆EC小鼠在PNX后的肺功能、重量体积均受损,免疫染色和流式细胞术分析表明,PNX后Tgm2i∆EC/i∆EC小鼠肺泡上皮足细胞和EC增殖受阻,纤维组织扩张加剧等,表明EC中TGM2的单胺化对PNX后的肺再生至关重要。为明确TGM2依赖性单胺化调控肺再生的分子机制,作者通过KEGG分析发现铁死亡相关基因在Tgm2i∆EC/i∆EC小鼠中显著富集。铁死亡抑制蛋白1(FSP1)和谷胱甘肽过氧化物酶4(GPX4)是铁死亡的关键抑制因子,FSP1/ GPX4的下调会导致脂质过氧化并引发铁死亡,作者发现FSP1和GPX4在Tgm2i∆EC/i∆EC小鼠的肺EC中表达降低,表明TGM2依赖性单胺化可以抑制肺再生过程中EC的铁死亡。然后,作者将内皮特异性过表达FSP1的AAV-VEC-Tie-Fsp1病毒注射到Tgm2i∆EC/i∆EC小鼠肺中,以恢复肺内皮Fsp1的表达,发现可逆转Tgm2i∆EC/i∆EC小鼠被抑制的肺再生。为测试铁死亡对TGM2阻断引起的肺再生的抑制作用,作者在小鼠EC中敲入Gpx4,经TGM2抑制剂(TGM2i)处理的Gpx4 EC敲入小鼠在PNX治疗后,肺功能、肺泡结构和细胞繁殖的恢复能力均增强。这些结果表明,肺EC中TGM2依赖性单胺化可通过抑制铁死亡而维持肺再生。
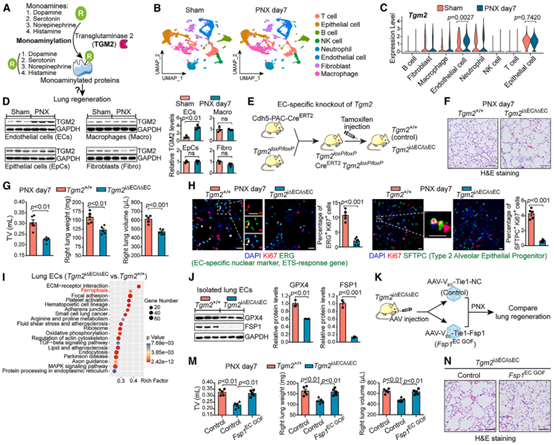
图1. 内皮细胞TGM2通过抑制铁死亡维持肺再生
作者为了揭示TGM2介导的神经递质修饰在EC中的抗铁死亡作用。使用不同的神经递质处理人脐静脉内皮细胞(HUVEC),发现只有多巴胺能显著抑制铁死亡,此外,使用TGM2i处理能够消除多巴胺对铁死亡的抑制作用及对GPX4的上调作用,表明多巴胺的抗铁死亡功能依赖于TGM2介导的多巴胺化修饰。由于多巴胺的生物合成需要3,4-二羟基苯丙氨酸脱羧酶(DDC),因此,作者使用了DDC缺失的小鼠模型探究多巴胺对肺再生的作用,发现DDC单倍体缺失小鼠在PNX模型中表现出肺功能、重量、体积和肺泡结构的恢复受阻,上皮细胞和EC增殖受阻以及纤维组织过度扩增等问题。为筛选EC中的多巴胺化蛋白,作者通过化学蛋白质组学、点击化学技术并与液相色谱-串联质谱分析相结合,鉴定出TPI1是TGM2介导的多巴胺化靶标之一。为检测内皮TGM2是否介导了TPI1多巴胺化,作者发现Tgm2i∆EC/i∆EC小鼠的肺EC中,TPI1的多巴胺化消失,表明体内TPI1的多巴胺化依赖于EC表达的TGM2。随后,作者通过体外和体内实验分别研究了TPI1对铁死亡的作用,发现敲低HUVEC中的Tpi1阻断了多巴胺抑制铁死亡和上调GPX4的作用,而过表达Tpi1可保护HUVEC免受铁死亡影响。在体内实验中,作者使用AAV-VEC-Tie-shTpi1特异性敲低肺EC中的Tpi1后(Tpi1EC KD),向小鼠注射pro-DA,发现AAV-VEC-Tie-shTpi1抑制了PNX后肺功能的恢复以及肺中的细胞繁殖。而注射pro-DA 会增强了Gpx4i∆EC/i∆EC 小鼠的肺再生能力,但Tgm2i∆EC/i∆EC小鼠没有改善。综上所述,TPI1作为TGM2介导的多巴胺化靶点抑制肺再生EC中的铁死亡。
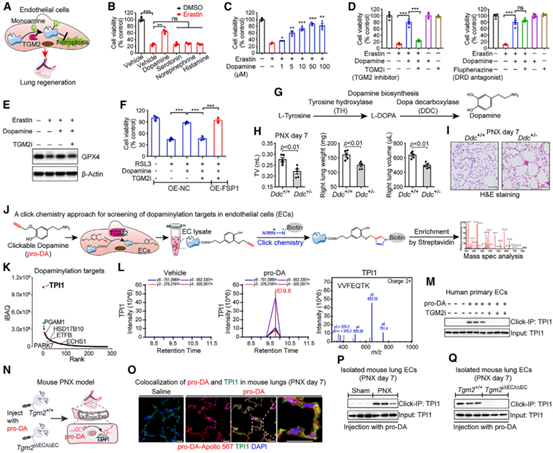
图2. TPI1是TGM2介导的多巴胺化靶标,可抑制再生肺内皮细胞中的铁死亡
接着,作者继续探究了TPI1调控铁死亡的具体机制。作者发现恢复肺EC中FSP1的表达可逆转肺切除Tpi1EC KD小鼠受损的肺再生能力,且内皮细胞和肺泡上皮祖细胞增殖增强,纤维扩增减少,EC选择性敲入GPX4可以改善Tpi1EC KD小鼠肺再生,这些发现表明TPI1的多巴胺化抑制ECs中的铁死亡以维持肺再生。随后,作者发现多巴胺能够增加TPI1催化DHAP转化为GAP的活性,而其他神经递质则没有此作用,TGM2i或多巴胺合成抑制剂可以减弱这种转化作用。因此,作者假设TGM2对TPI1的多巴胺化作用介导了将DHAP转化为GAP的酶活性变化,并调节醚类磷脂的合成。作者通过对肺内皮细胞进行脂质组学分析,发现TGM2i增强了磷脂、磷脂酰胆碱及相关醚磷脂的生成,TPI1在HUVEC中的敲低也增加了醚磷脂的产生,这些磷脂在脂质过氧化后会引发铁死亡。此外,Tpi1EC KD小鼠在肺内皮细胞中表现出更高的脂质过氧化相关的铁死亡水平,而这种铁死亡可以通过过表达Fsp1逆转,这些结果表明,TPI1的多巴胺化调节醚磷脂的产生,以防止PNX后脂质过氧化和铁死亡。
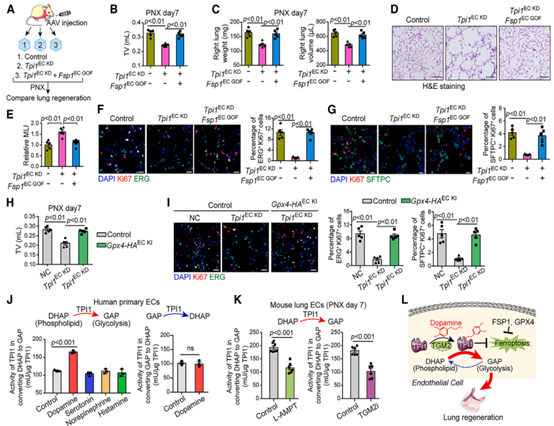
图3. TPI1的多巴胺化调节醚磷脂合成,以调节再生肺内皮细胞中的脂质过氧化和铁死亡
由于蛋白质中的谷氨酰胺(Q)已被证明是TGM2的转氨位点,因此,作者使用LC-MS/MS分析了TPI1中谷氨酰胺残基的修饰情况,结果显示,在TGM2存在的情况下,TPI1通过Q65残基被多巴胺化修饰,在Q65处观察到分子量改变,将TPI1中的Q65突变为谷氨酸会阻断TGM2对其进行多巴胺化修饰,此结果表明,多巴胺共价修饰了TPI1的Q65残基,进而增强了其将DHAP转化为GAP的活性。为验证Q65多巴胺化的TPI1对肺的保护作用,作者纯化出了能特异性识别Q65多巴胺化TPI1的多克隆抗体,WB分析显示,在肺切除的Tgm2i∆EC/i∆EC小鼠的肺EC细胞中,Q65位点的TPI1多巴胺化被消除。作者把TPI1的Q65残基突变为谷氨酸(Tpi1EC Q65E)来研究Q65多巴胺化TPI1的体内功能,作者将AAV-VEC-Tie-shTpi1和AAV-VEC-Tie-Q65E Tpi1共同注射到小鼠肺中,WB分析显示,在PNX后,Tpi1EC Q65E小鼠肺EC中TPI1的Q65多巴胺化修饰被消除,同时肺泡再生被抑制,肺纤维扩张增强。综上所述,肺内皮细胞TPI1中Q65多巴胺化避免了脂质过氧化和铁死亡,从而维持了肺再生。
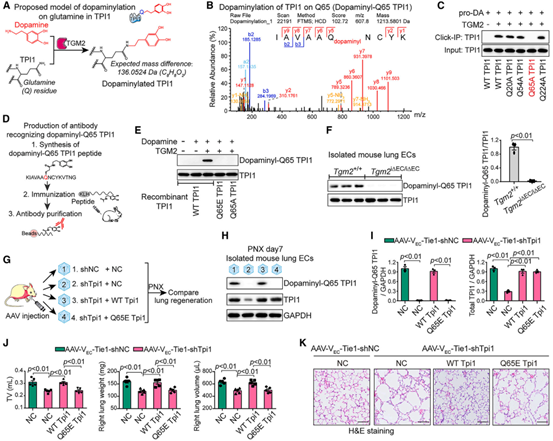
图4. TPI1中Q65的多巴胺化修饰可减少促纤维化的铁死亡并促进肺再生
随后,作者对Q65多巴胺化修饰影响TPI1催化活性和方向的机制进行了探究。作者发现,Tpi1EC Q65E小鼠在PNX后,肺EC中DHAP生成增加,GAP生成减少,同时糖酵解减少,与铁死亡相关的磷脂过氧化作用增强,表明TPI1中Q65的多巴胺化选择性地提高了其将脂质前体DHAP转化为糖酵解组分GAP的活性,从而平衡了脂质和葡萄糖代谢。接着,作者采用分子动力学模拟方法分析发现TPI1中Q65的多巴胺化会导致TPI1的构象变化,从而加强了催化残基与DHAP而非GAP的接触,与未修饰的TPI1相比,多巴胺化后的TPI1与DHAP的距离更短,尤其是催化残基H96和K14与DHAP之间的距离缩短,相比之下,催化残基与GAP之间的距离不受多巴胺化的影响,此外,多巴胺化还优化了催化残基E166相对于DHAP的角度,使得E166的位置更加有利于催化DHAP。综上表明,多巴胺化TPI1的构象变化优化了TPI1与DHAP的相互作用,减少了与GAP的相互作用,这有助于促进糖酵解并减少脂质过氧化,平衡了脂质/葡萄糖代谢,最终抑制铁死亡,促进肺再生。
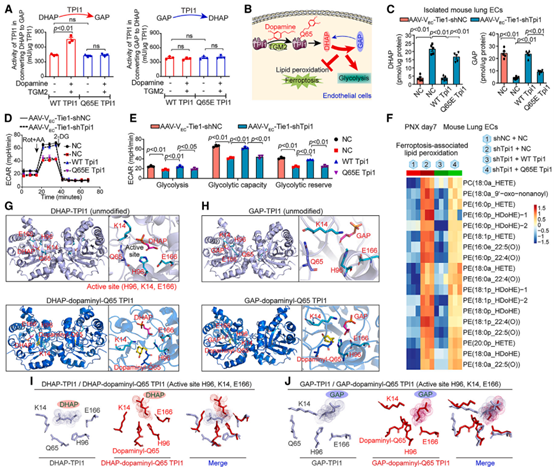
图5. TPI1中Q65残基的多巴胺化增强了其催化残基与DHAP而非GAP之间的接触,引导酶促反应将DHAP转化为GAP
由于阻断肺EC中的TPI1多巴胺化会增加纤维扩张,作者评估了TPI1多巴胺化在肺再生中的抗纤维化作用。发现PNX后,Tgm2i∆EC/i∆EC小鼠肺胶原沉积和纤维反应增强,Tpi1EC KD小鼠肺纤维化反应增强,而内皮细胞过表达Fsp1则使Tpi1EC KD肺纤维化反应减弱。由于血管内皮细胞会产生多种旁分泌因子与血管周围细胞相互作用,因此,作者检测了PNX后Tgm2i∆EC/i∆EC和Tpi1EC KD小鼠肺EC中旁分泌因子的分化表达,其中肝细胞生长因子(HGF)是肺EC中分化程度最高的基因,PNX后,Tgm2i∆EC/i∆EC和Tpi1EC KD小鼠肺EC中HGF蛋白减少,肺内皮细胞中HGF的产生已被证明可刺激上皮细胞增殖并阻断纤维活化。为了检测HGF的作用,作者使用了Tgm2i∆EC/i∆EC和Tpi1EC KD小鼠的肺切片进行体外培养,发现培养基中的HGF蛋白水平下降,而在培养基中添加HGF可增加肺泡上皮祖细胞的增殖,并减弱了纤维化。这些结果表明,阻断再生肺内皮细胞中TPI1多巴胺化可诱导铁死亡,并重新编程内皮旁分泌因子/血管分泌因子如HGF的产生。
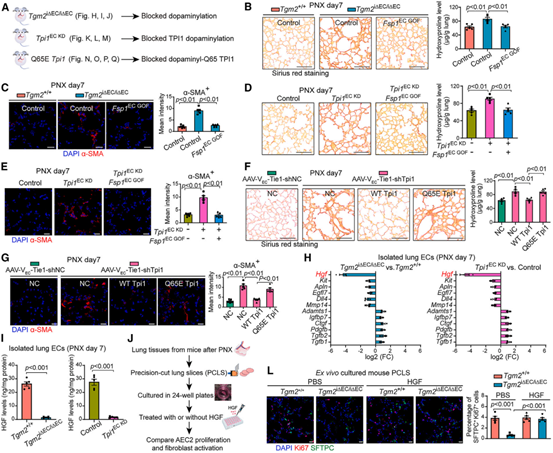
图6. 阻断EC中的TPI1多巴胺化会增强PNX后小鼠肺部的纤维化反应
由于阻断内皮TPI1的Q65残基多巴胺化会增强PNX后的纤维激活,作者进一步评估了Q65多巴胺化的TPI1在肺纤维化患者和小鼠肺纤维化模型中的作用。在肺纤维化患者的样本中,通过单细胞RNA测序分析,发现TGM2在纤维化肺内皮细胞中的表达减少,且与肺功能指标的恶化相关联,Q65多巴胺化的TPI1量也有所降低。在小鼠肺纤维化模型中,肺EC中TGM2和Q65多巴胺化的TPI1均降低,这些数据表明,TPI1的Q65位点经TGM2多巴胺化后可减轻肺纤维化。然后,作者在肺纤维化小鼠模型中检测Tgm2i∆EC/i∆EC和
Tpi1
EC KD和Tpi1
EC Q65E小鼠的相关表型,发现Tgm2
i∆EC/i∆EC和Tpi1
EC KD肺纤维化增强,而恢复肺EC中Fsp1表达使肺纤维化减少,另外,Tpi1
EC Q65E小鼠中肺纤维化增强,表明内皮TPI1的抗纤维化作用主要依赖于Q65位点的多巴胺化。然后,作者在肺纤维化小鼠模型中测试了pro-DA的作用,向肺纤维化小鼠注射pro-DA可提高肺EC细胞中Q65多巴胺化的TPI1含量,且改变了损伤肺EC中与再生相关的血管分泌基因的表达,这表明多巴胺化影响了血管周围细胞与内皮细胞之间的连接,而pro-DA在Tgm2
i∆EC/i∆EC小鼠中不存在这种作用,表明pro-DA主要通过内皮细胞中表达的TGM2介导的多巴胺化来减轻肺纤维化。
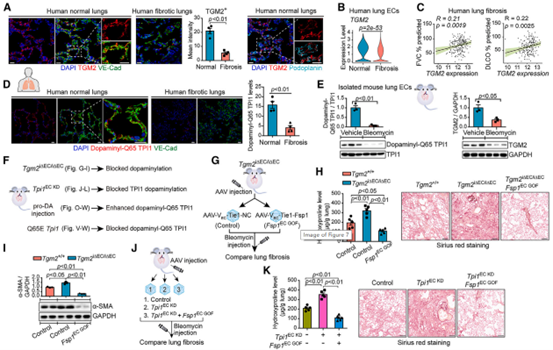
图7. 内皮细胞TPI1 Q65残基的多巴胺化可减少铁死亡,以减轻肺纤维化
综上所述,该研究发现了肺内皮细胞中TGM2介导的TPI1 Q65位点多巴胺修饰可减少脂质过氧化从而抑制铁死亡,进而有效抑制肺纤维化的进展。此外,通过深入解析肺再生的调控机制,不仅为器官纤维化的再生疗法确定了潜在的治疗靶点,也为未来开发针对血管微环境的药物提供了科学依据。









 8007
8007










