胰腺导管腺癌(PDAC)是最致命的恶性肿瘤之一,大多数患者在被诊断出时病情已至晚期,化疗是晚期PDAC的主要治疗策略。白蛋白结合型紫杉醇联合吉西他滨方案(AG方案)是胰腺癌的主要化疗方案,而原发性耐药极大影响了AG方案的效果。经典的PI3K/Akt通路是多种肿瘤相关信号通路中的核心通路,异常激活可大大促进PDAC的化疗耐药性,使用靶向该通路的抑制剂组合是提高患者化疗敏感性的潜在策略。然而,众多化疗联合PI3K/Akt通路抑制剂治疗策略所显示出的治疗有效性和安全性仍然十分有限,探索安全可靠的PI3K/Akt通路抑制剂对于提高PDAC化疗敏感性非常重要。

福建医科大学胜利临床医学院福州大学附属省立医院陈实教授团队携手福州大学应用基因组学研究所陈鲤群,在《Molecular Cancer》(IF:27.7)发表题为《Sulindac (K-80003) with nab-paclitaxel and gemcitabine overcomes drug-resistant pancreatic cancer》的研究报告,通过对AG耐药患者来源类器官(PDO)、PDAC细胞系和异种移植模型进行体内和体外实验验证,发现PI3K/Akt通路的异常激活在AG治疗方案的耐药性中起着核心作用,确定了一种经典的抗炎药——舒林酸衍生物K-80003——与AG方案联合使用可显著增强化疗敏感性;筛选出可预测AG化疗敏感性的Hsa_circ_0030292(cFAM124A)作为生物标记物,其作用机制为cFAM124A通过增强组织蛋白酶L(CTSL)表达及蛋白活性,刺激RXRα水解,形成大量截短的tRXRα,激活PI3K/Akt通路并促进PDAC中的GEM耐药。在研究中,作者使用汉恒生物提供的cFAM124A过表达慢病毒和过表达质粒在PDAC细胞系PATU8988T和MiaPaCa-2中实现了cFAM124A的外源性过表达。

接下来,我们一起看看本研究的主要结果。
首先,作者通过活检获取了20例晚期不可切除PDAC患者的肿瘤组织,通过肿瘤影像学和肿瘤标志物糖类抗原19-9(CA19-9)表达的变化将患者分为AG敏感组和AG耐药组,对这些组织的RNA-seq数据进行KEGG分析,结果显示PI3K/Akt信号通路具有最显著变化、且具有最高的富集得分。接着,作者构建了AG耐药PDO模型(PDO-AGR组)和AG敏感PDO模型(PDO-AGS组)以验证两组间的药物耐受性差异。PDO-AGS组在AG处理下显示出更低的IC50值和较低的细胞死亡率,对PDO的RNA测序分析显示PI3K/Akt通路在PDO-AGR组异常激活,表明PI3K/Akt通路的异常激活在PDAC的AG耐药中起关键作用。
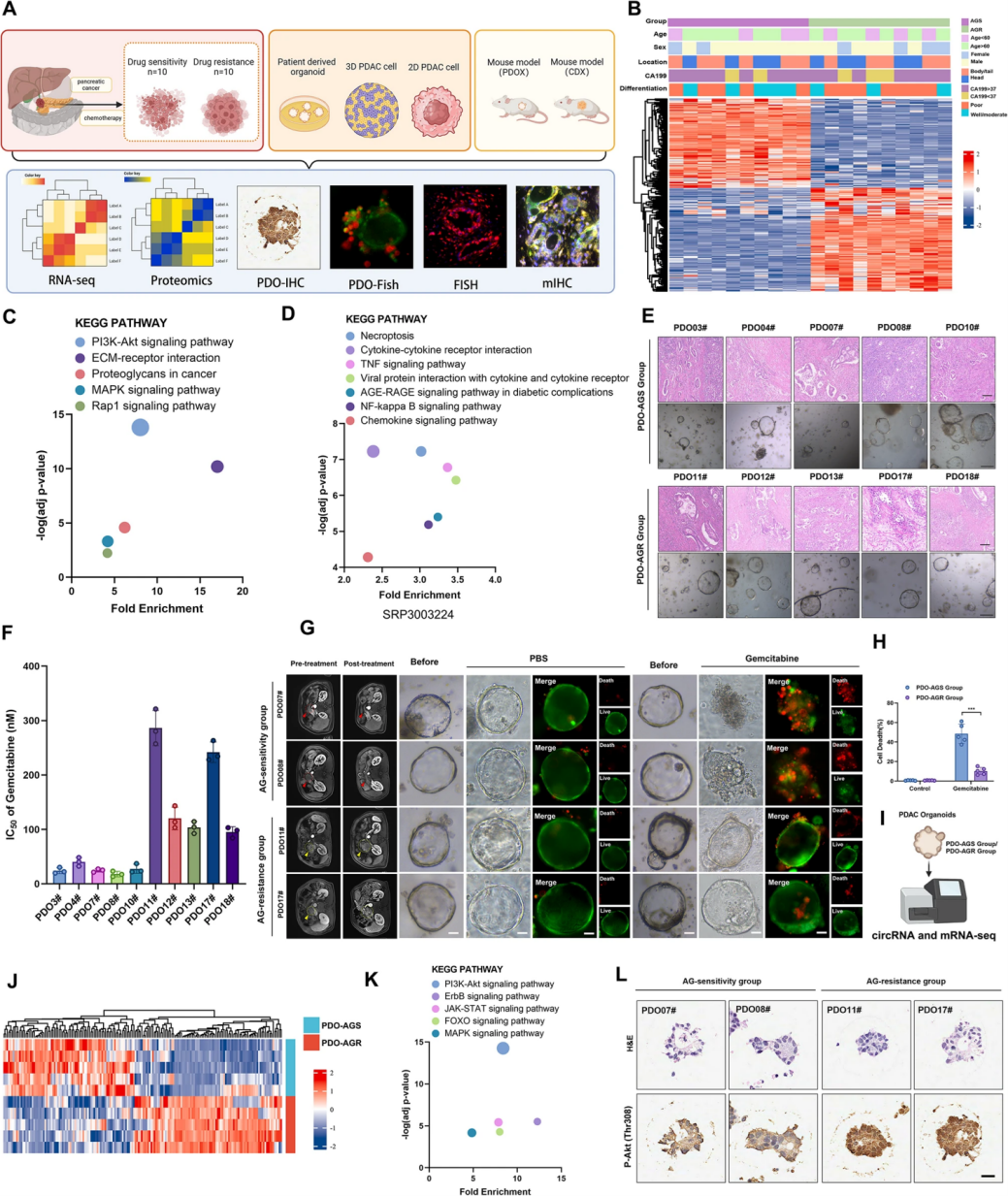
图1 PI3K/Akt通路在AG耐药PDAC中起关键作用
由于现有的PI3k/Akt抑制剂具有一定毒性和副作用,不适用于临床应用,而作者团队此前发现已投入临床使用的抗炎药舒林酸K-80003是有效的I3K/Akt通路抑制剂,因此,本次研究探索了K-80003是否具有克服在PDO-AGR组AG耐药性的潜力。在5个AG耐药的PDO模型中,K-80003使其中3个模型(PDO11#,PDO17#,和PDO18#)恢复AG化疗敏感性,AG与K-80003联合治疗显著降低了该PDO-AGR组别的细胞存活率。此外,在建立的PDO原位胰腺癌小鼠模型(PDOX模型)中,与单独使用AG方案相比,联合使用K-80003和AG方案显著减小了肿瘤体积,并延长了相应的PDOX模型的中位生存期。作者使用内部、外部PDO数据库随机选择的耐药PDO构建PDOX模型,AG联合K-80003治疗同样使得部分PDOX模型表现出化疗敏感性,这证明K-80003是一种有效的AG化疗敏感增强剂,但只有一半患者具有治疗效果,表明需要找到合适的反应预测标志物来实现患者分层治疗。
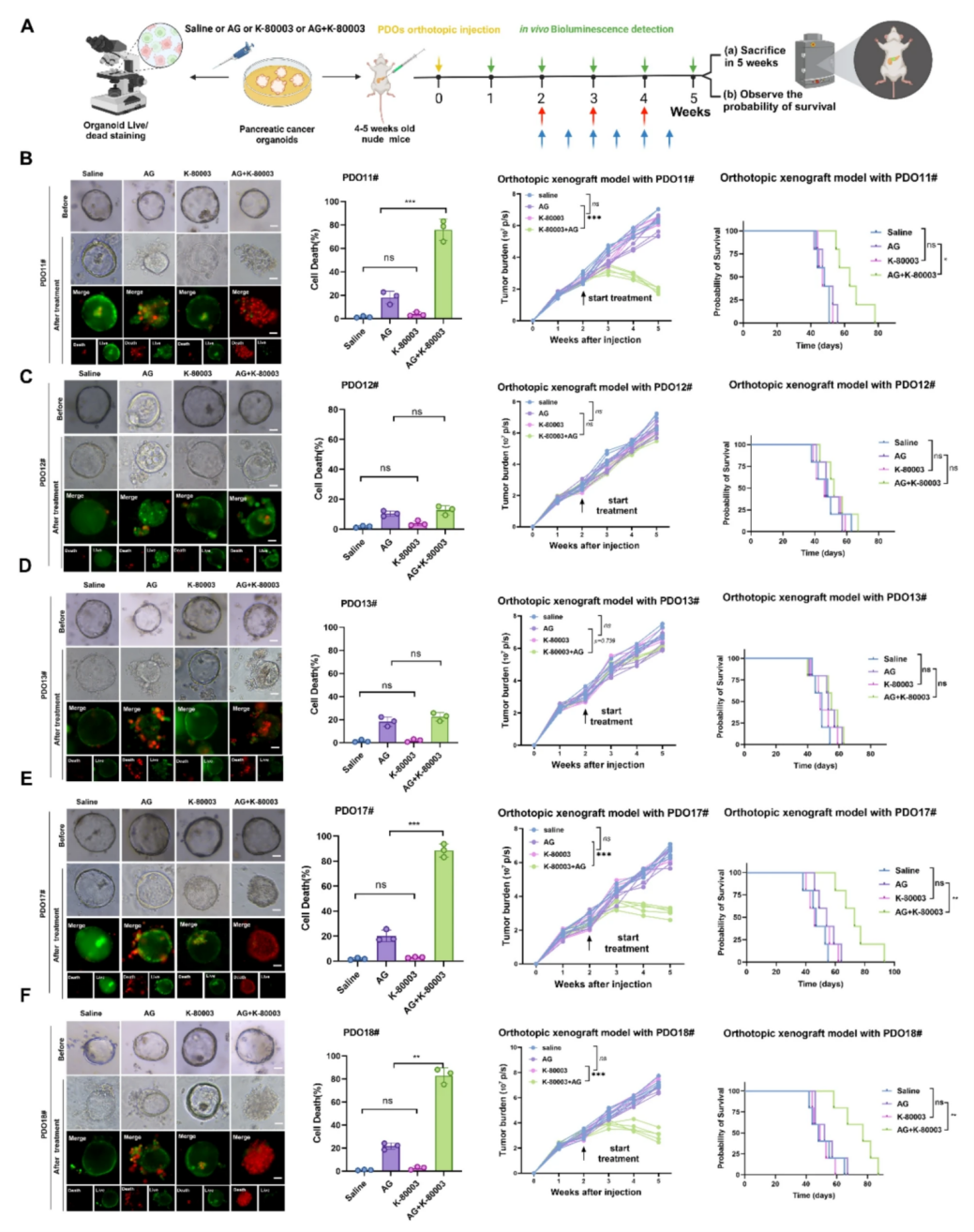
图2 K-80003可提高部分AG耐药PDAC患者肿瘤类器官对AG化疗的敏感性
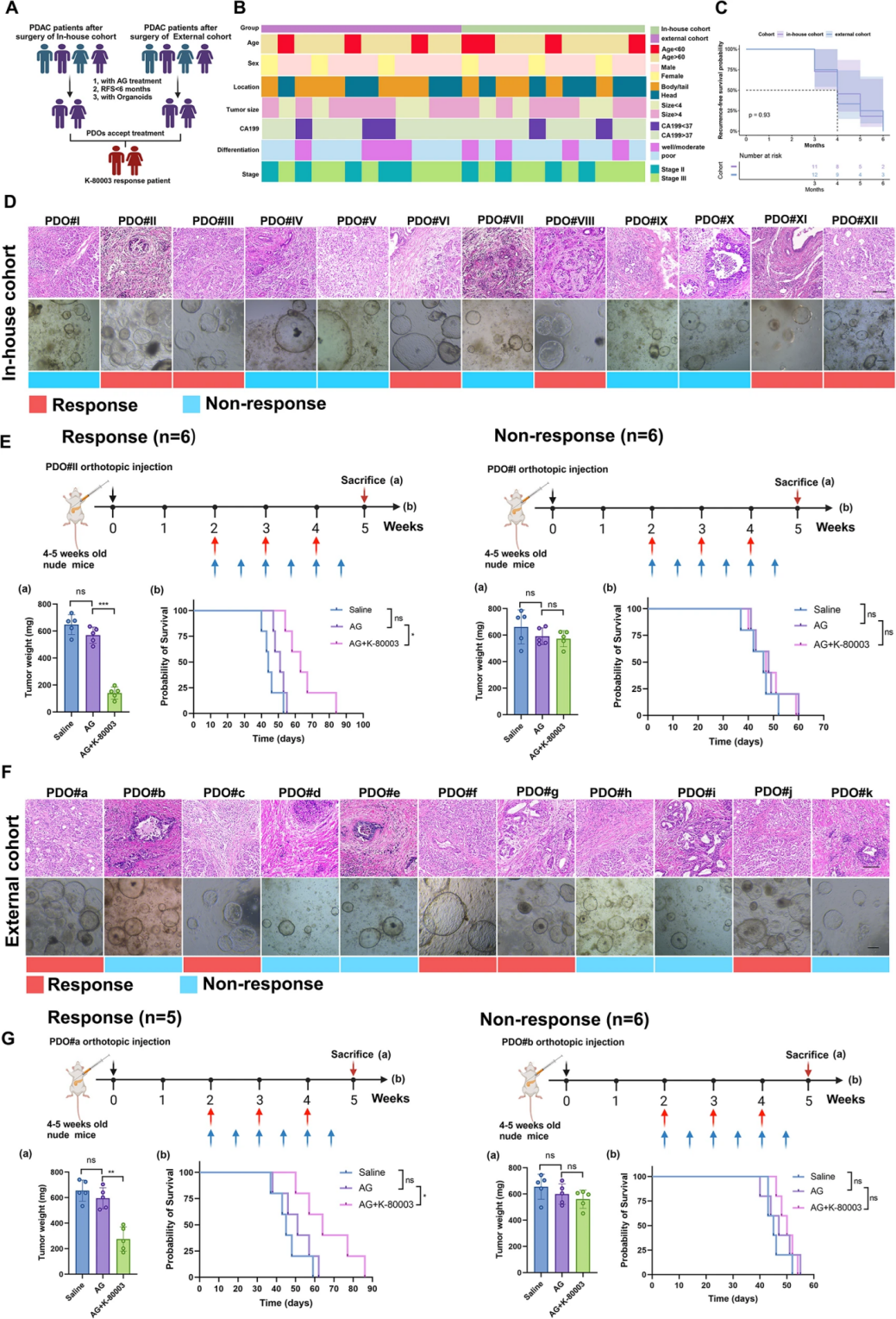
图3 K-80003可增强AG耐药临床PDOX模型样本对AG化疗的敏感性
为了探索与K-80003提高AG化疗敏感性能力相关的潜在circRNA标志物,作者对PDO样本的RNA-Seq数据进行分析,筛选鉴定出46个差异表达的circRNA,并对上调/下调变化最为显著的前5个分子进行PCR验证。通过circRNA与PI3K/AKT通路之间关联的基因集富集分析(GSEA)和Pearson相关性分析确定4个circRNA,分别于PDAC细胞系PATU8988T中进行过表达,验证其与吉西他滨(GEM,AG方案中的主要成分之一)和K-80003给药条件下的细胞化疗敏感性是否相关,并进行3D肿瘤微球凋亡染色测定,确定了cFAM124A与GEM敏感性相关度最高。接受GEM化疗的PDAC患者肿瘤组织中的IHC染色和FISH结果显示,与GEM敏感组相比,GEM耐药组中的cFAM124A和p-Akt(Thr308)表达显著上升,二者的变化呈正相关。此外,cFAM124A表达与PDAC患者的的生存率密切相关。以上结果表明只有cFAM124A而非其他上调的circRNA,与GAM相关且可预测K-80003治疗效果。
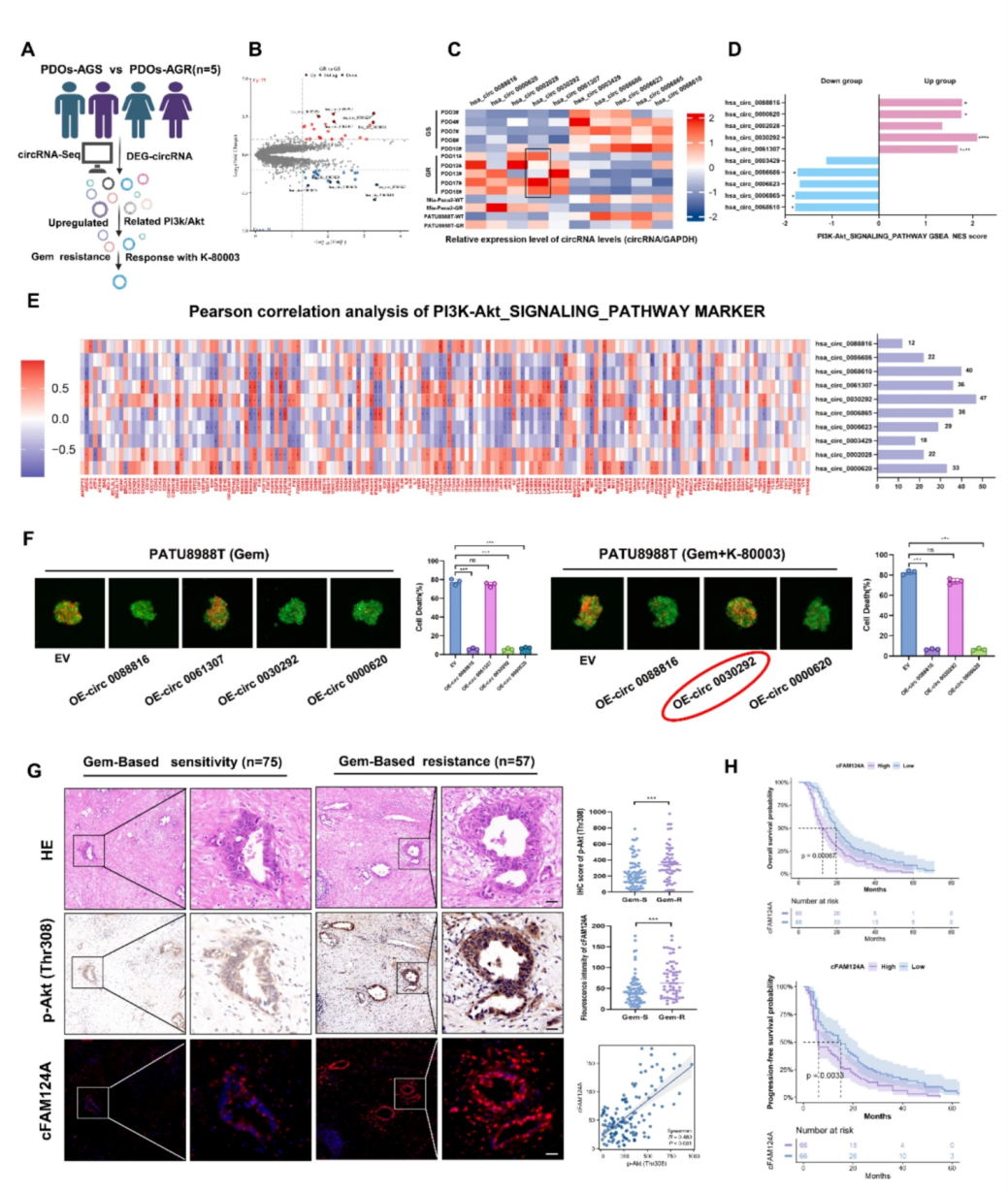
图4 cFAM124A是K-80003促进PDAC中AG化疗敏感的潜在分子
PI3K/Akt抑制剂K-80003可通过阻止tRXRα与PI3K的p85α亚基结合来抑制PI3K/Akt信号的激活,然而tRXRα的截短蛋白特性使得其不适合作为预测性生物标志物,于是,作者进一步验证p-Akt(Thr308)p-Akt(Ser473)和cFAM124A组织表达差异与K-80003的相关性,以及其对比预测K-80003治疗应答的能力。mIFS染色和IHC染色结果显示PDAC患者K-80003反应组和K-80003无反应组组织中的cFAM124A丰度差异显著,癌症组织中的cFAM124A、p-Akt(Thr308)和p-Akt(Ser473)的表达水平高于癌旁组织中的表达水平。肿瘤相对于癌旁组织的倍数变化值显示,cFAM124A在反应组和非反应组之间具有良好的区分能力,而P-Akt(Thr308)和P-Akt(Ser473)识别效果远不如cFAM124A,这表明cFAM124A是一种实用有效的生物标志物。
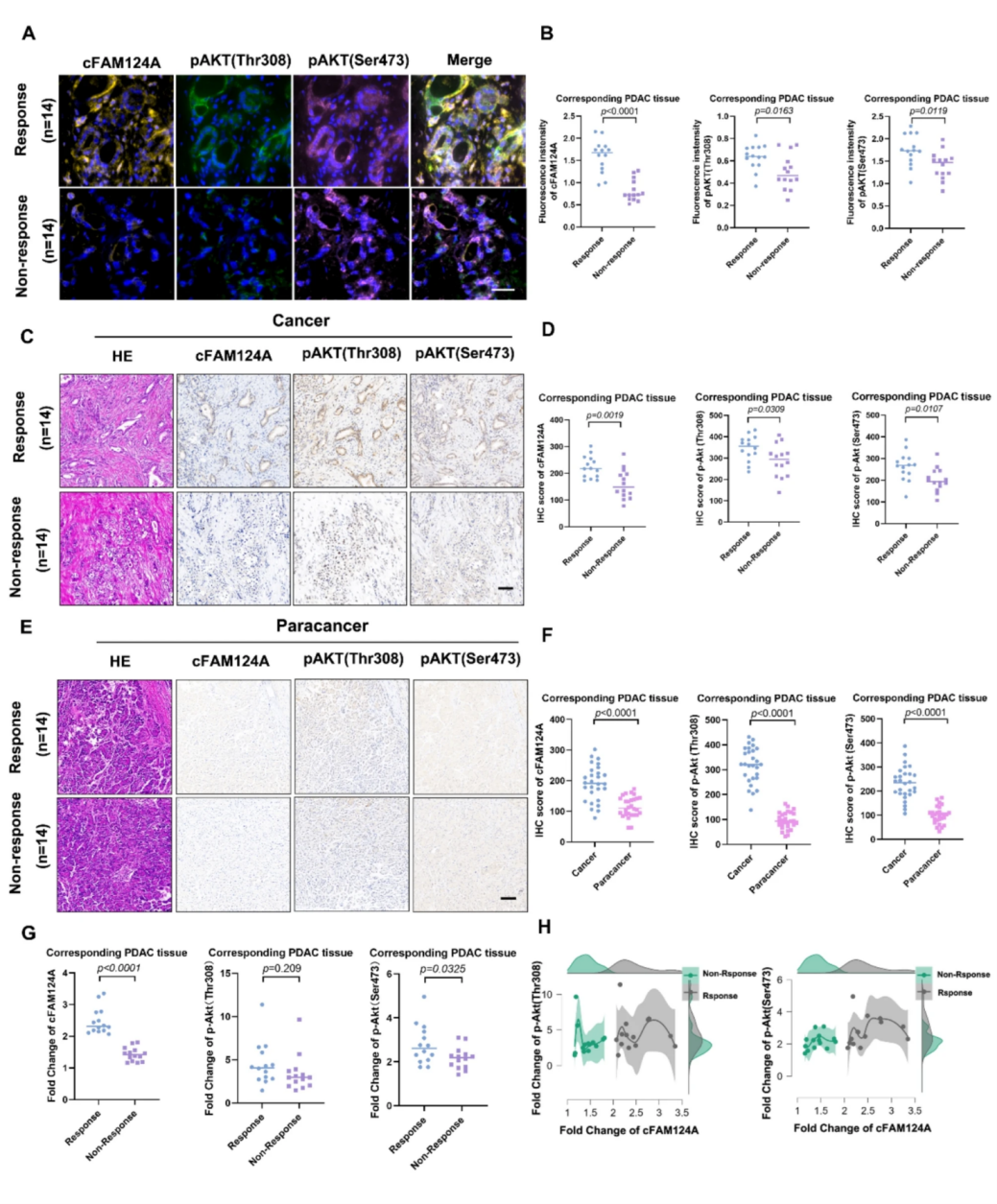
图5 cFAM124A是预测K-80003促进AG敏感性能力的最佳标志物
为了探究cFAM124A作为预测K-80003药物反应标志物的机制,作者在PATU8988T和MiaPaCa-2细胞中过表达cFAM124A,通过WB检测发现cFAM124A的过表达减弱了PDAC细胞中GEM诱导的细胞凋亡水平,促进了PI3K/Akt信号通路的过度激活,PI3K泛抑制剂(PI3Ki,copanlisib)则抑制了这些变化。CCK-8测定、3D肿瘤微球凋亡染色实验和克隆形成实验等体外实验,以及皮下异种移植模型体内实验结果表明,copanlisib的使用恢复了cFAM124A过表达所导致的GEM耐药性。然而,copanlisib可导致小鼠体重减轻,K-80003对小鼠体重无显著影响,这表明K-80003具有更好的安全性。随后,作者进行了蛋白组学分析和WB检测以探究cFAM124A所调控的下游蛋白质表达变化,结果显示cFAM124A过表达显著激活PI3K/Akt通路,抑制RXRα蛋白表达和降解,最终促进tRXRα(截短型RXRα)的产生。由于RXRα蛋白水解产生的截短蛋白tRXRα与PI3K的p85α亚基相互作用可激活PI3K/Akt通路,作者推测cFAM124A通过tRXRα激活PI3K/Akt通路,导致PDAC中的GEM耐药,而K-80003可以逆转这种作用。
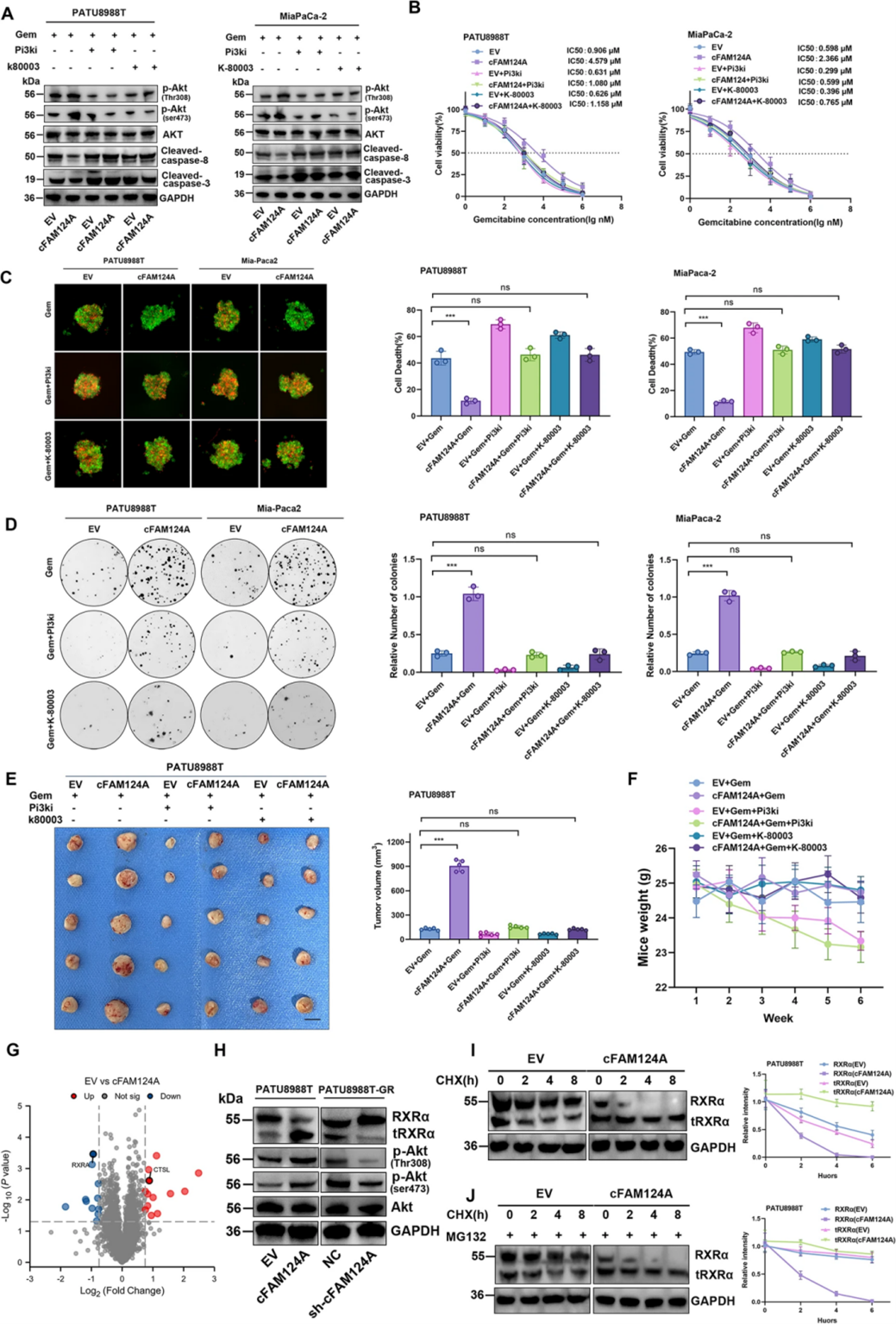
图6 FAM124A通过tRXRα激活PI3K/Akt通路引起PDAC的GEM耐药
接下来,作者研究了cFAM124A促进RXRα水解的机制,发现CTSL酶抑制剂(Z-FY-CHO),而不是m-钙蛋白酶抑制剂(PD150606),可恢复由cFAM124A过表达引起的RXRα下降和tRXRα增加,而cFAM124A可促进活性CTSL累积。作者通过数据库预测、RNA pulldown-MS鉴定和RIP-qPCR等分析,证实了cFAM124A和胰岛素样生长因子2RNA结合蛋白2(IGF2BP2)之间存在相互作用。通过RNA pulldown和RNA反义纯化(RAP)测定结果证实了IGF2BP2和CTSL mRNA之间的相互作用,cFAM124A过表达增加了IGF2BP2和CTSL mRNA之间的相互作用,导致CTSL mRNA的降解速度变慢。为了确认IGF2BP2介导的CTSL mRNA直接功能域(识别m6A的功能域,KH4)的稳定和依赖cFAM124A的间接促进功能域(cFAM124A结合的功能域,KH1)的功能,作者进一步构建IGF2BP2截断体,证明了cFAM124A结合IGF2BP2,以m6A依赖的方式稳定CTSL mRNA,从而发挥桥接作用。cFAM124A支架位点的突变在mRNA和蛋白水平上完全逆转了cFAM124A过表达诱导的CTSL表达上调,而只能部分逆转cFAM124A过表达引起的CTSL活性、tRXRα水平、p-Akt/Akt表达量以及GEM耐药性增加。CTSL的敲低则完全逆转了cFAM124A过表达导致的tRXRα/PI3K/ATK通路和GEM耐药性变化,这表明cFAM124A还通过其他机制影响CTSL酶活性。
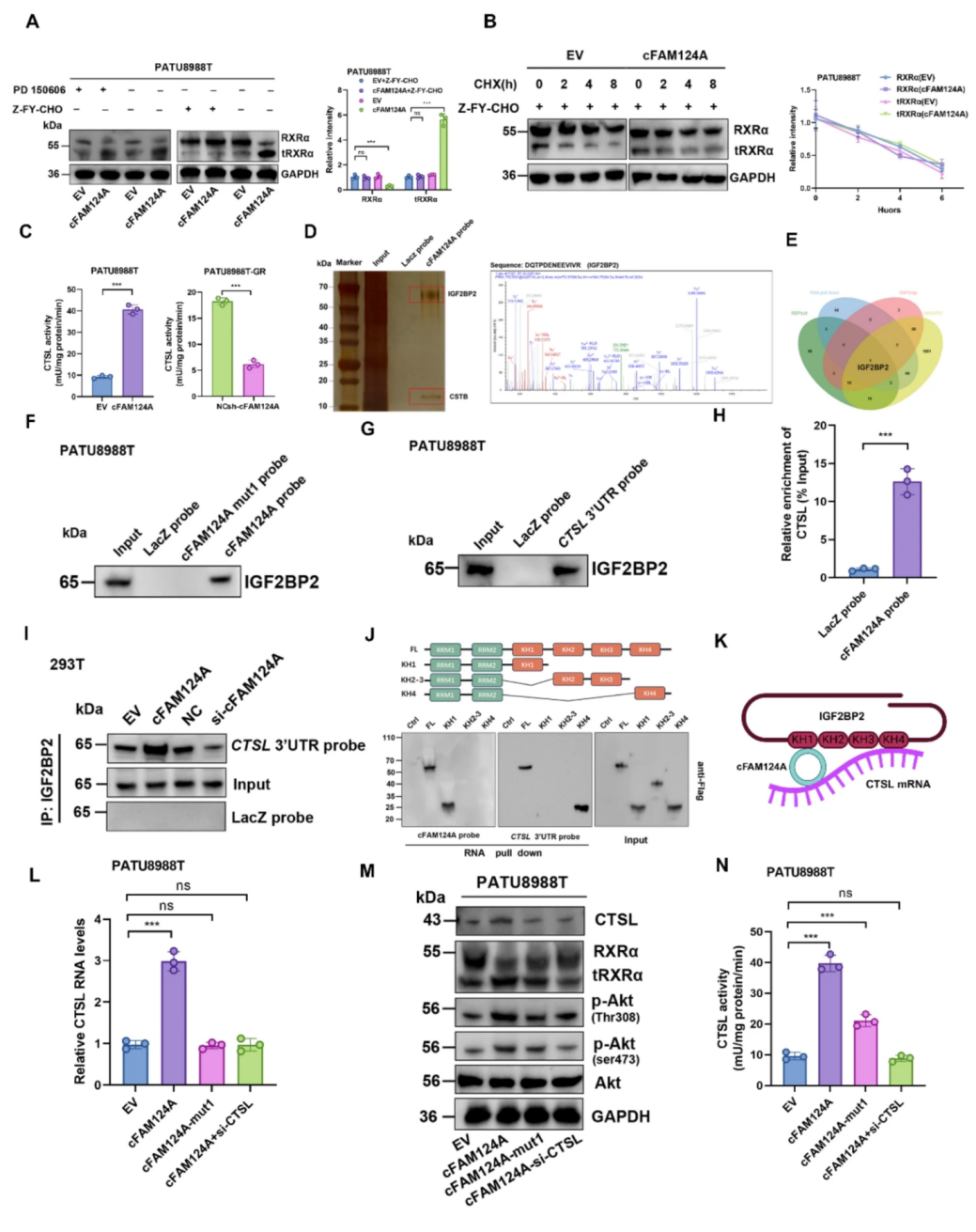
图7 cFAM124A通过桥接作用上调CTSL酶表达,并促进tRXRα蛋白水平的增加
CTSL的活性主要受胱抑素A(CSTA)和胱抑素B(CSTB)调节,为了探究cFAM124A是否也通过该通路调控CTSL活性,作者进行了RNA pulldown-MS分析,并通过免疫荧光、RIP和RNA pulldown实验证明了FAM124A和CSTB之间存在相互作用,cFAM124A的表达显著抑制了CTCL-CSTB相互作用。使用catRAPID数据库预测结合位点,并构建去除了与CSTB结合的竞争位点的cFAM124A截短序列质粒,结果显示在cFAM124A的76-210-nt区域被截断时,circRNA探针捕获的CSTB蛋白显著减少,同时CSTB与CTSL结合的增加。此外,WB、CTSL酶活性检测、3D肿瘤微球染色和集落形成实验的结果显示竞争位点的缺失减弱了cFAM124A过表达所引起的tRXRα的增加、PI3K/Akt通路激活的异常升高和CTSL活性,而cFAM124A支架位点和竞争位点的同时突变则逆转了这些变化。
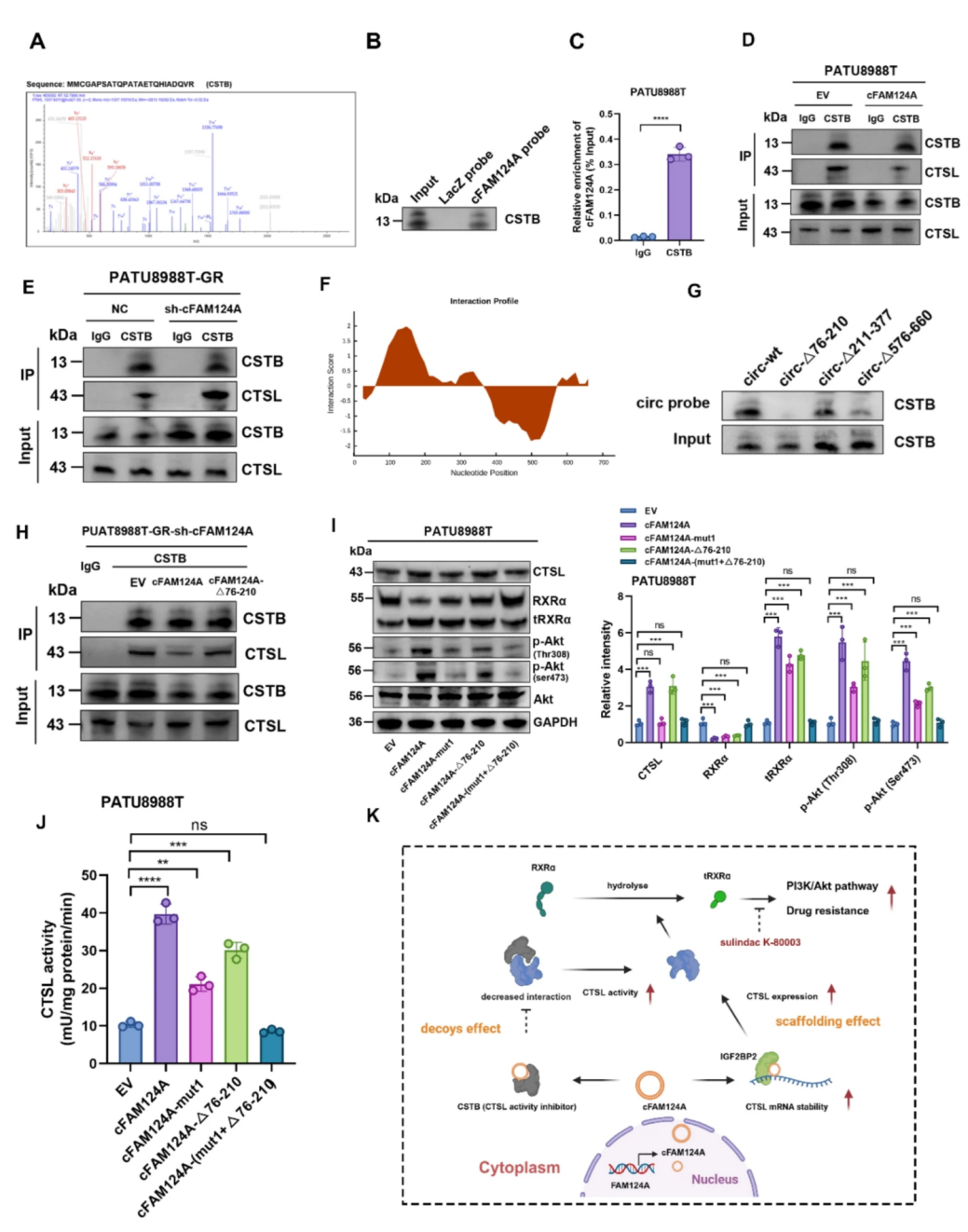
图8 cFAM124A通过与CSTB竞争性结合增强CTSL酶活性
综上所述,本研究探索了PDAC对AG方案致敏的新治疗策略,证实了抑制PI3K/Akt的成熟药物K-80003的临床潜力,发现cFAM124A通过与IGF2BP2蛋白、CTSL mRNA结合来形成更稳定的复合物,并通过与CSTB蛋白结合来负向调节CTSL蛋白酶的活性,是预测PDAC中K-80003克服AG化疗耐药能力的最佳生物标志物。









 7494
7494










