哺乳动物昼夜节律系统通过协调代谢与行为(如进食—禁食周期)来维持组织稳态,其核心分子钟(如BMAL1、CLOCK等)在肝脏中高度活跃,并受外周环境信号(如进食时间)调控。肝脏生物钟通过调节分解与合成代谢通路,影响能量稳态和损伤修复。然而,其在协调再生相关代谢需求(如核苷酸合成)中的作用尚未明确。生物钟紊乱(如基因缺失或昼夜节律失调)是否通过代谢失衡加剧再生障碍?本研究解析了肝生物钟通过调控磷酸戊糖途径(PPP)的活性影响核苷酸合成的分子机制,进而揭示了生物钟失调导致肝再生障碍的代谢根源,并探索了基于代谢重编程(如激活PPP限速酶G6PDX)或术前干预(如间歇性禁食)的促修复策略。

2025年1月,中国药科大学钱民先教授、王中原教授以及深圳大学刘宝华教授作为共同通讯作者在Nature Metabolism(IF 18.9)期刊发表题为“The hepatic clock synergizes with HIF-1α to regulate nucleotide availability during liver damage repair”的研究论文。基于核苷酸的可用性对DNA复制和修复是至关重要的。本研究发现,肝脏中的昼夜节律通过控制PPP的活性,支持核苷酸的从头合成,以满足DNA合成的需求。研究显示,通过基因操作或非正常喂养时间扰乱肝生物钟会损害雄性小鼠的PPP活性,导致核苷酸失衡。这种缺失不仅引发DNA复制应激,限制肝切除后的再生能力,还会导致基因毒素诱导的肝细胞衰老和STING信号依赖性炎症。从机制上看,分子钟激活因子BMAL1与缺氧诱导因子-1α(HIF-1α)协同调控PPP限速酶葡萄糖-6-磷酸脱氢酶(G6PDX)的转录,而G6PDX在肝脏再生过程中表达增强。过表达G6PDX可以恢复Bmal1或HIF-1α缺失肝脏的再生能力。此外,通过基因手段或术前间歇性禁食(IF)提高G6PDX表达,显著促进了正常小鼠的肝脏修复。因此,本研究结果强调了肝生物钟的生理重要性,并提出了一种有前景的促再生策略。
在本研究中,汉恒生物有幸为作者提供了AAV8-shG6PDX和AAV8-Flag-G6PDX,成功调控了小鼠肝脏中G6PDX的表达。
下面,我们一起来了解具体的研究内容:
1、肝脏时钟紊乱损害肝脏再生过程中的细胞周期进程
为探究肝生物钟在损伤后肝细胞增殖中的作用,作者建立了2/3肝切除(PH)模型,发现PH术后第2天,核心钟基因Bmal1和Clock表达显著上调,而抑制因子(如Cry1、Per2)下降。若通过肝特异性Bmal1敲除(LBKO)或4天限时进食(DRF)扰乱肝生物钟后,再生指数(肝/体重比)及Ki-67+增殖肝细胞数量均显著降低,且肝损伤标志物ALT/AST持续升高,证实肝生物钟完整性是急性损伤后再生的必要条件。随后,BrdU标记(可标记增殖细胞)显示LBKO和DRF组肝细胞DNA复制较对照组延迟约12小时,且复制峰未及时消退。而PCNA(S期关键因子)表达在对照组术后24小时达峰值后下降,实验组却持续高表达,提示S期进程受阻并产生复制应激。综上所述,作者认为肝生物钟通过协调DNA复制进程保障再生效率,其紊乱会导致S期延迟及复制应激,最终限制修复能力。
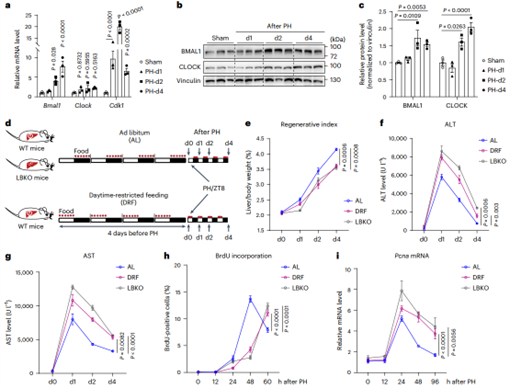
图1. 肝脏时钟紊乱导致S期进展失败并损害肝脏再生
2、肝脏时钟紊乱损害对肝细胞增殖至关重要的戊糖磷酸途径活性
接下来,作者进一步探究了LBKO和DRF影响肝脏再生的机制。通过代谢组学分析发现,在肝生物钟紊乱模型(LBKO和DRF小鼠)中,PPP关键中间代谢物(6-PG、S7P、R5P)的节律性被显著破坏,且PPP限速酶G6PDX的活性和mRNA表达节律消失。随后,通过同位素示踪实验发现Bmal1缺失肝细胞的PPP代谢通量显著降低,而糖酵解终产物乳酸生成增加,提示葡萄糖代谢向糖酵解倾斜。接下来,作者验证了PPP在肝再生中的作用,在肝脏切除术后,肝细胞G6PDX活性和表达显著升高,而Bmal1敲除完全阻断这一响应。最后,通过AAV介导的G6pdx敲低显著延缓了肝切除后的修复进展,以上结果表明PPP对肝脏再生不可或缺。
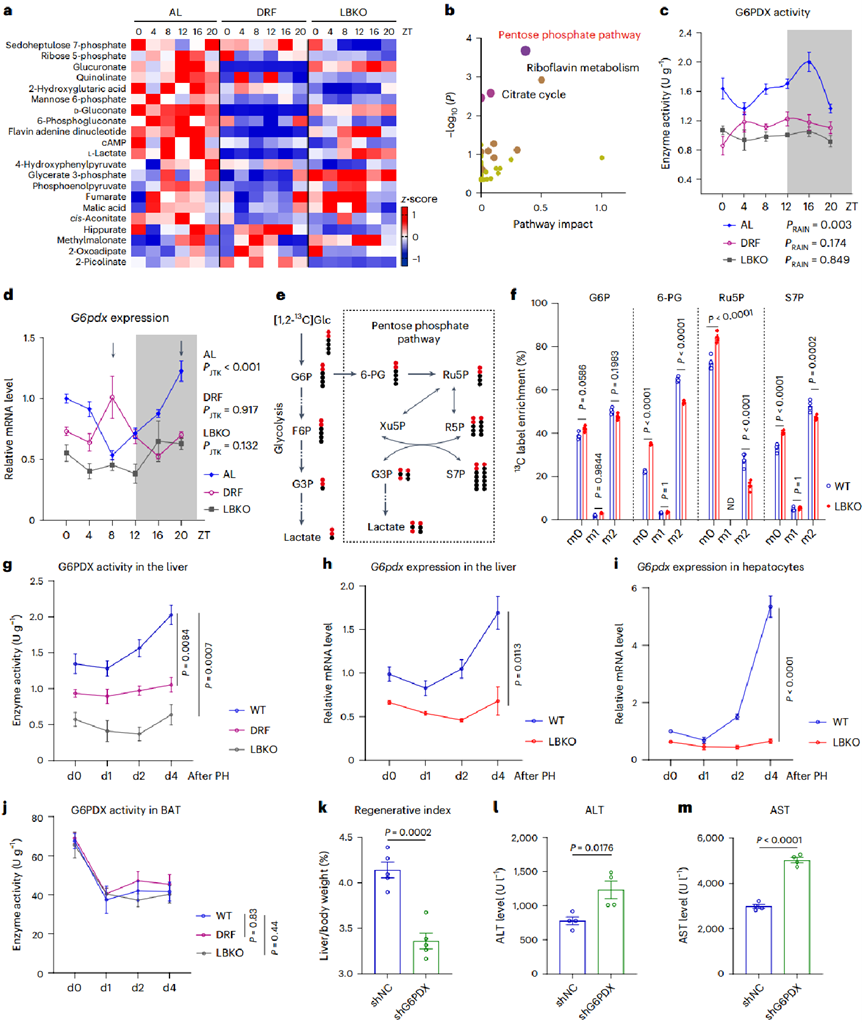
图2. 肝生物钟紊乱损害PPP的节律性和活性
3、肝生物钟的紊乱通过氧化应激升高和核苷酸短缺来限制肝细胞增殖
PPP是产生胞质烟酰胺腺嘌呤二核苷酸磷酸盐(NADPH)的主要途径,在正常肝脏中NADPH水平呈现昼夜节律性,而生物钟紊乱模型中该节律消失且总量显著降低。进行肝切除术后,对照组NADPH和谷胱甘肽(GSH)水平升高以应对氧化应激,而生物钟紊乱组两者水平持续偏低。但给予强效抗氧化剂N-乙酰半胱氨酸(NAC)可显著降低活性氧(ROS)和8-氧-鸟嘌呤水平,部分恢复LBKO和DRF小鼠的肝再生能力。这些结果表明,PPP功能障碍引起的ROS不平衡在一定程度上阻碍了LBKO和DRF小鼠的肝脏再生过程。
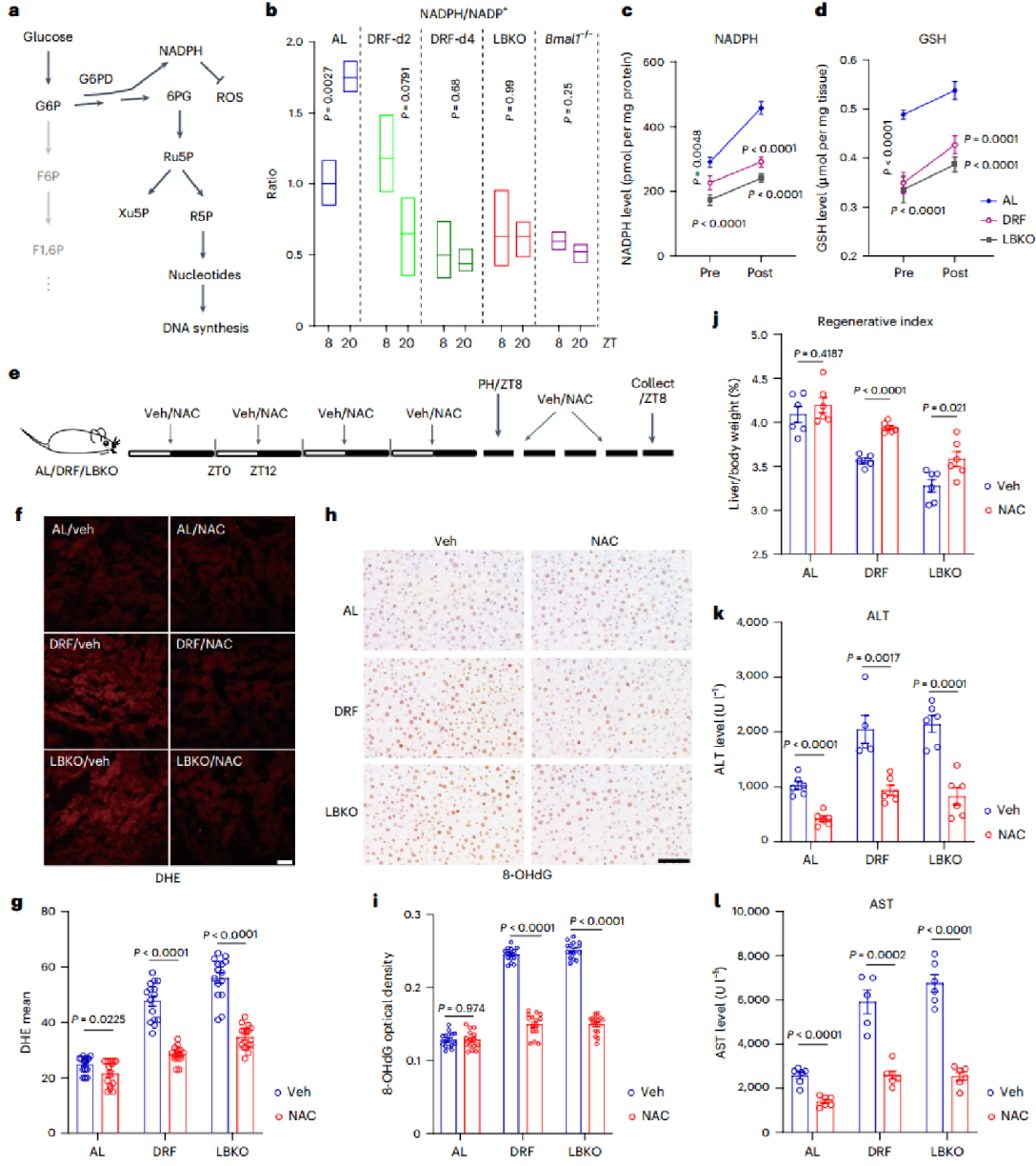
图3. 肝生物钟破坏中氧化应激升高部分导致再生缺失
此外,PPP的R5P产物是核苷酸合成的前体,肝脏中G6pdx敲低显著降低了核苷酸含量。同时,代谢组学分析显示抑制G6PDX或Bmal1缺失均可显著降低原代肝细胞中戊糖磷酸的水平。经检测,野生型(WT)小鼠PH术后核苷酸池扩大,而LBKO小鼠因PPP活性受损无法响应此需求,但过表达G6pdx可显著恢复LBKO小鼠肝脏中(脱氧)核苷酸三磷酸[(d) NTPs]的水平。因此,作者认为Bmal1缺失细胞中的PPP功能障碍导致了核苷酸短缺。而核苷酸供应不足会在增殖细胞中引发复制应激,这在Bmal1敲除或G6PDX抑制剂(6-AN)处理的肝细胞中得到验证:其DNA复制叉延伸速度显著减慢,提示复制应激。生物钟紊乱小鼠在PH术后由于复制应激检查点(CHK1、RPA32)的持续激活,导致损伤细胞的积累,但在自由进食的小鼠中几乎检测不到。类器官培养实验表明Bmal1缺失的肝细胞在体外生长受限,但联合抗氧化剂NAC与核苷酸补充或过表达G6PDX可显著恢复其增殖能力。综上所述,由肝生物钟介导的PPP缺失会导致ROS积累和核苷酸短缺,从而损害肝脏再生。
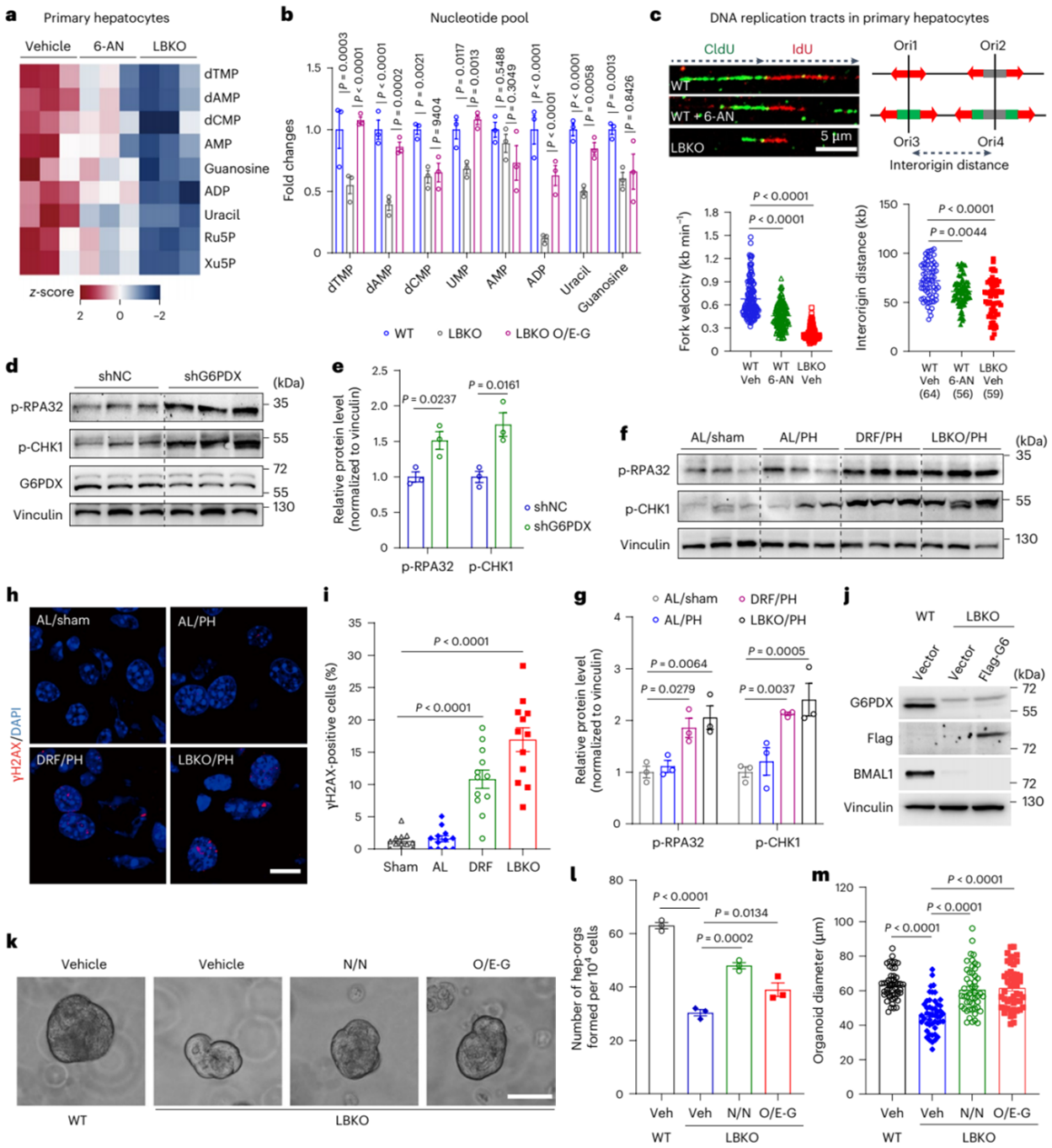
图4. PPP功能障碍引起的核苷酸短缺导致Bmal1缺失细胞的复制应激和生长缺失
4、肝脏BMAL1协同HIF-1α控制G6pdx的表达
随后,作者进行了核心分子钟是否以转录方式直接控制PPP活性的验证。Bmal1缺失型小鼠原代肝细胞中G6pdx mRNA水平的显著降低表明G6pdx是一个由分子钟靶向的PPP标志基因。通过双荧光素酶和染色质免疫沉淀(ChIP)分析实验发现,BMAL1与HIF-1α形成二聚体,作为核心转录激活因子通过优先结合G6pdx启动子的(A/T)ACG TGT(E’-box)基序来调控其表达。而BMAL1和HIF-1α在肝脏中呈现节律性结合,且肝切除术后结合增强,两者共表达可显著激活E’-box驱动的荧光素酶活性,且敲低任一方均会削弱另一方的结合能力。以上结果表明,肝脏BMAL1与HIF-1α协同控制G6pdx的转录。

图5. BMAL1与HIF-1α协同控制小鼠肝脏中G6PDX表达的节律和丰度
5、增加PPP活性促进损伤后的肝脏修复
为了确定增强PPP活性是否可以挽救LBKO小鼠的肝再生,作者使用AAV系统实现小鼠肝脏中G6PDX过表达。结果表明G6PDX过表达显著改善了LBKO小鼠的再生指数(肝/体重比)、降低了其ALT/AST水平,并恢复了Ki-67+增殖肝细胞的比例;同时,逆转了HIF-1α敲除小鼠的术后再生障碍,证实HIF-1α通过调控G6PDX影响再生;G6PDX过表达也加速了WT小鼠肝脏功能修复以及核苷酸池的扩增。此外,既往研究表明BMAL1活性可以通过禁食调整,因此,作者在PH手术之前对小鼠进行IF处理。ChIP和染色质组分测定结果表明,IF通过增强BMAL1和HIF-1α的DNA结合能力,显著上调G6pdx表达及酶活性。经IF预处理的小鼠PH术后核苷酸池扩大,再生能力得以改善。而敲除Bmal1或G6pdx完全消除了IF的促再生效果,证明其发挥功能依赖BMAL1-G6PDX轴。
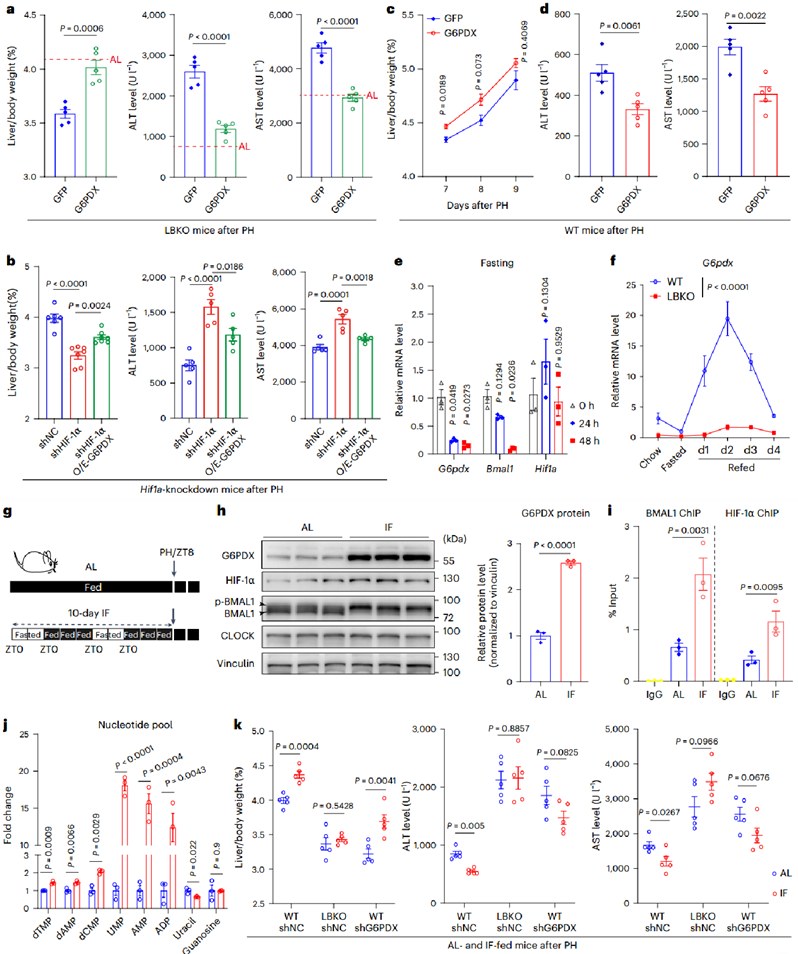
图6. 增强PPP活性有助于肝脏再生
6、Bmal1缺失通过核苷酸短缺削弱DNA修复,从而驱动肝细胞衰老和炎症
接着,作者验证了Bmal1缺失是否影响同源重组(HR)的修复效率。结果表明Bmal1敲低或G6PDX抑制均会导致细胞HR修复效率下降,而补充核苷酸或过表达G6PDX可部分恢复。DNA损伤剂(辐射或百草枯)处理后,在Bmal1缺失的肝脏中观察到不可修复DNA损伤的积累,导致衰老肝细胞比例升高,而过表达G6PDX显著促进了损伤DNA的清除,并减少了衰老肝细胞的数量。在机制上,Bmal1缺失肝脏中的持续性DNA损伤导致cGAS-STING通路的异常激活,最终导致炎症衰老相关的分泌表型,而抑制STING则可减少肝脏炎症。以上结果表明,DNA损伤的积累驱动了STING这一DNA传感通路的激活,从而将PPP缺失与LBKO小鼠的肝脏炎症及衰老相关联。
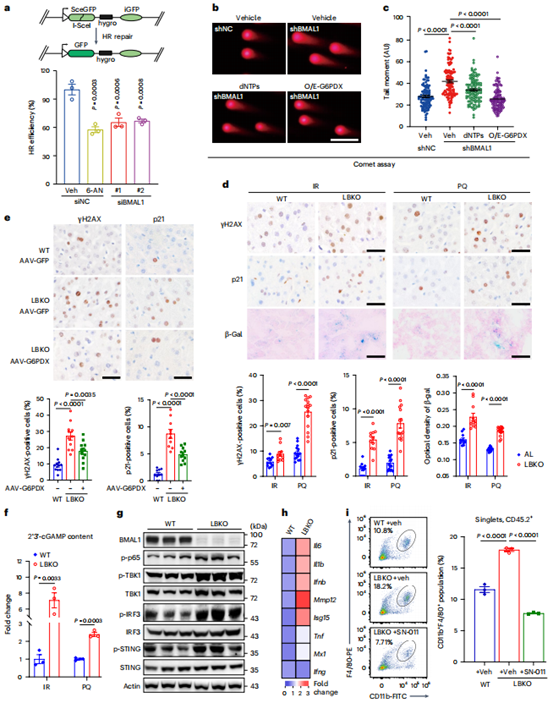
图7. 核苷酸短缺阻碍Bmal1缺失细胞中的DNA修复,导致衰老和炎症
综上所述,肝生物钟(BMAL1)与HIF-1α协同调控G6PDX节律性表达,进而维持核苷酸合成与抗氧化能力。而肝脏生物钟紊乱会导致氧化应激和核苷酸失衡,最终导致肝脏再生障碍。本研究结果表明,靶向G6pdx(基因干预)或术前IF可优化核苷酸代谢,进而改善肝修复与抗炎能力,是潜在的促再生策略。本研究首次揭示生物钟通过代谢—免疫交叉调控组织稳态,为肝损伤、衰老及炎症性疾病提供多靶点干预思路。
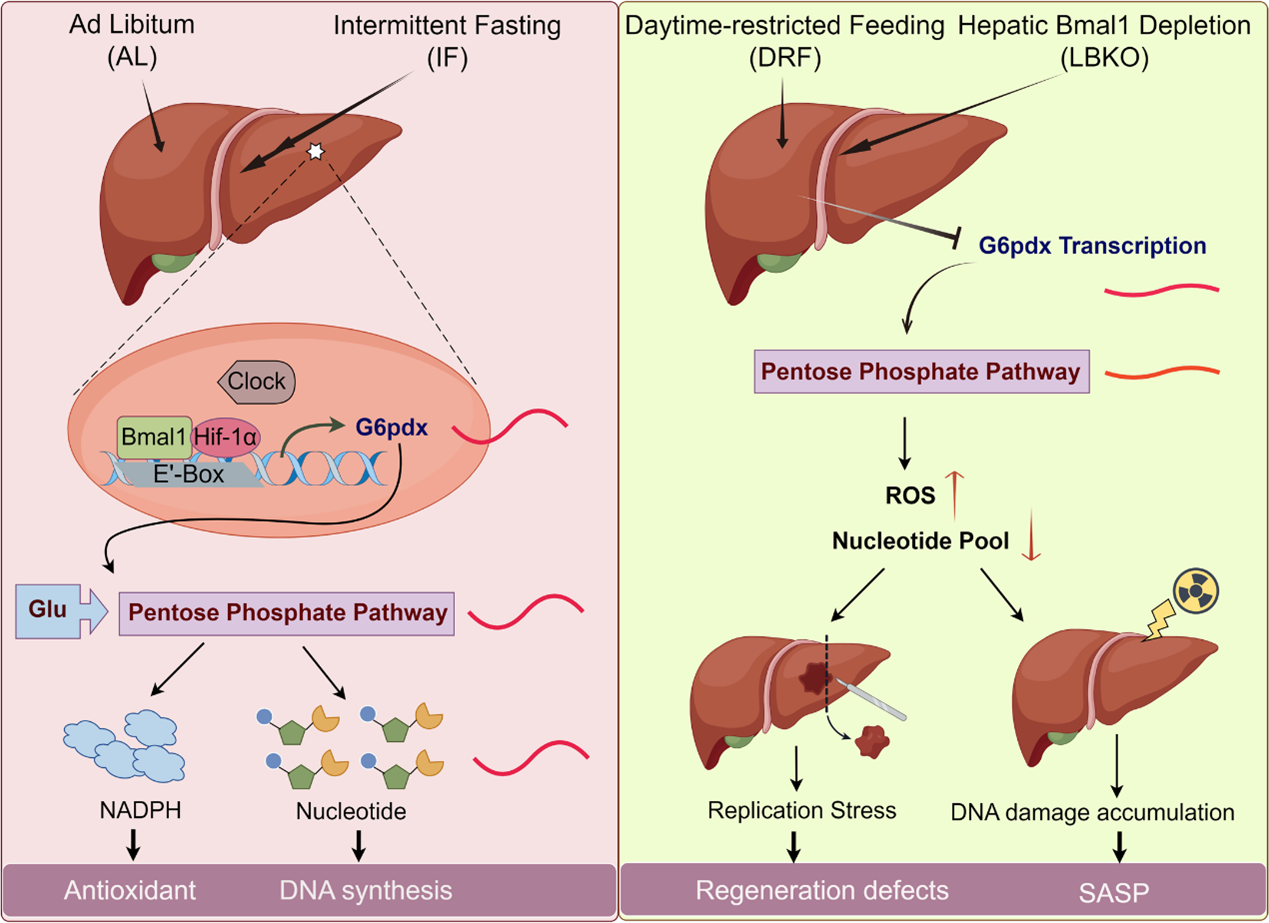
图形摘要. 肝生物钟介导肝脏再生的作用机制









 7231
7231










